IMPORTANT
NOTICE
This document provides information for an application for Illumina technology that has
been demonstrated internally and may be of interest to customers. This information is
provided as‐is and is not an Illumina product and is not accompanied by any rights or
warranties. Customers using or adapting this information should obtain any licenses
required and materials from authorized vendors. Illumina products mentioned herein are
for research use only unless marked otherwise. While customer feedback is welcomed, this
application is not supported by Illumina Technical Support and Field Application Scientists.
Part # 15044223 Rev. B
Page 1
16S Metagenomic Sequencing Library
Preparation
Preparing 16S Ribosomal RNA Gene Amplicons for the
Illumina MiSeq System
Introduction 2
16S Library Preparation Workflow 5
Amplicon PCR 6
PCR Clean‐Up 8
Index PCR 10
PCR Clean‐Up 2 13
[Optional] Validate Library 15
Library Quantification, Normalization, and Pooling 16
Library Denaturing and MiSeq Sample Loading 17
MiSeq Reporter Metagenomics Workflow 20
Supporting Information 21
Introduction
Metagenomic studies are commonly performed by analyzing the prokaryotic 16S ribosomal
RNA gene (16S rRNA), which is approximately 1,500 bp long and contains nine variable
regions interspersed between conserved regions. Variable regions of 16S rRNA are frequently
used in phylogenetic classifications such as genus or species in diverse microbial
populations.
Which 16S rRNA region to sequence is an area of debate, and your region of interest might
vary depending on things such as experimental objectives, design, and sample type. This
protocol describes a method for preparing samples for sequencing the variable V3 and V4
regions of the 16S rRNA gene. This protocol can also be used for sequencing other regions
with different region‐specific primers. This protocol combined with a benchtop sequencing
system, on‐board primary analysis, and secondary analysis using MiSeq Reporter or
BaseSpace, provides a comprehensive workflow for 16S rRNA amplicon sequencing.
Workflow Summary:
1 Order amplicon primers–The protocol includes the primer pair sequences for the V3 and
V4 region that create a single amplicon of approximately ~460 bp. The protocol also
includes overhang adapter sequences that must be appended to the primer pair
sequences for compatibility with Illumina index and sequencing adapters. Illumina does
not sell these primers. They must be ordered from a third party. See Amplicon Primers,
on page 3 for more information on amplicon primers.
2 Prepare library–The protocol describes the steps to amplify the V3 and V4 region and
using a limited cycle PCR, add Illumina sequencing adapters and dual‐index barcodes
to the amplicon target. Using the full complement of Nextera XT indices, up to 96
libraries can be pooled together for sequencing.
3 Sequence on MiSeq–Using paired 300‐bp reads, and MiSeq v3 reagents, the ends of each
read are overlapped to generate high‐quality, full‐length reads of the V3 and V4 region
in a single 65‐hour run. The MiSeq run output is approximately > 20 million reads and,
assuming 96 indexed samples, can generate > 100,000 reads per sample, commonly
recognized as sufficient for metagenomic surveys.
4 Analyze on MSR or BaseSpace–The Metagenomics workflow is a secondary analysis
option built into the MiSeq Reporter (on‐system software) or available on BaseSpace
(cloud‐based software). The Metagenomics Workflow performs a taxonomic
classification using the Greengenes database showing genus or species level
classification in a graphical format.
This protocol can be used to sequence alternative regions of the 16S rRNA gene and for other
targeted amplicon sequences of interest. When using this protocol for amplicon sequencing
other than 16S rRNA, use the Generate FASTQ Workflow (secondary analysis option). For
more information, see MiSeq Reporter Metagenomics Workflow, on page 20.
DISCLAIMER
The information in this Illumina Demonstrated Protocol is being provided as a courtesy; in
some cases reagents are required to be purchased from non‐authorized third‐party suppliers.
Illumina does not guarantee nor promises technical support for the performance of our
products used with reagents purchased from a non‐authorized third‐party supplier.
Introduction
Page 2
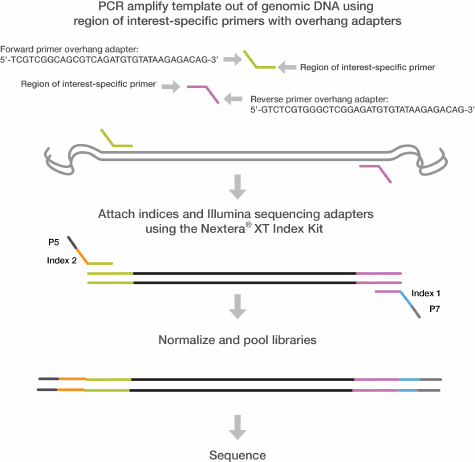
Figure 1 16S V3 and V4 Amplicon Workflow
User‐defined forward and reverse primers that are complementary upstream and downstream of the
region of interest are designed with overhang adapters, and used to amplify templates from genomic
DNA. A subsequent limited‐cycle amplification step is performed to add multiplexing indices and
Illumina sequencing adapters. Libraries are normalized and pooled, and sequenced on the MiSeq
system using v3 reagents.
Amplicon Primers
• The gene‐specific sequences used in this protocol target the 16S V3 and V4 region. They
are selected from the Klindworth et al. publication (Klindworth A, Pruesse E, Schweer T,
Peplles J, Quast C, et al. (2013) Evaluation of general 16S ribosomal RNA gene PCR
primers for classical and next‐generation sequencing‐based diversity studies. Nucleic
Acids Res 41(1).) as the most promising bacterial primer pair. Illumina adapter
overhang nucleotide sequences are added to the gene‐specific sequences. The full length
primer sequences, using standard IUPAC nucleotide nomenclature, to follow the protocol
targeting this region are:
16S Amplicon PCR Forward Primer = 5'
TCGTCGGCAGCGTCAGATGTGTATAAGAGACAGCCTACGGGNGGCWGCAG
16S Amplicon PCR Reverse Primer = 5'
GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAGGACTACHVGGGTATCTAATCC
• This method can also be utilized to target other regions on the genome (either for 16S
with other sets of primer pairs, or non‐16S regions throughout the genome; ie any
amplicon). The overhang adapter sequence must be added to the locus‐specific primer
for the region to be targeted (Figure 1). The Illumina overhang adapter sequences to be
added to locus‐specific sequences are:
Forward overhang: 5’ TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG‐[locus‐
specific sequence]
Reverse overhang: 5’ GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG‐[locus‐
specific sequence]
Introduction
Page 3
• The following considerations are recommended for designing other locus‐specific
primers:
a Illumina recommends targeting regions that result in an amplicon that when
sequenced with paired‐end reads has at least ~50 bp of overlapping sequence in the
middle. For example, if running 2x300 bp paired‐end reads Illumina recommends
having an insert size of 550 bp or smaller so that the bases sequenced at the end of
each read overlap.
b The locus‐specific portion of primer (not including overhang sequence) must have a
melting temperature (Tm) of 60°–65°C. You can use online PCR primer sequence
analysis tools (e.g. http://www.idtdna.com/analyzer/Applications/OligoAnalyzer/) to
check the properties of primer designs. For the Tm calculation only, the gene‐specific
portion must be used in calculation. For hairpin and dimer calculations, the fully‐
assembled primer sequence (including the overhang) should be used.
c Illumina recommends using standard desalting purification when ordering oligo
primer sets.
NOTE
For more information on reagents used in the protocol, see Consumables and Equipment,
on page 21.
Introduction
Page 4
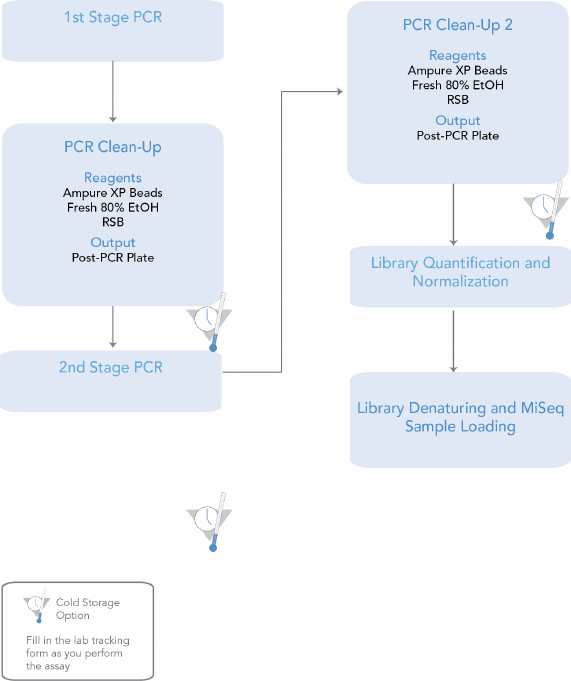
16S Library Preparation Workflow
The following diagram illustrates the workflow using the 16S Library Preparation Protocol.
Safe stopping points are marked between steps.
Figure 2 16S Library Preparation Workflow
16S Library Preparation Workflow
Page 5
Amplicon PCR
This step uses PCR to amplify template out of a DNA sample using region of interest‐
specific primers with overhang adapters attached. For more information on primer
sequences, see Amplicon Primers, on page 3.
Consumables
NOTE
For more information on consumables and equipment for this protocol see Consumables and
Equipment, on page 21.
Item Quantity Storage
Microbial Genomic DNA (5 ng/µl in 10 mM
Tris pH 8.5)
2.5 µl per sample ‐15° to ‐25°C
Amplicon PCR Reverse Primer (1 µM) 5 µl per sample ‐15° to ‐25°C
Amplicon PCR Forward Primer (1 µM) 5 µl per sample ‐15° to ‐25°C
2x KAPA HiFi HotStart ReadyMix 12.5 µl per sample ‐15° to ‐25°C
Microseal 'A' film
96‐well 0.2 ml PCR plate 1 plate
[Optional] Bioanalyzer chip (Agilent DNA 1000
kit catalog # 5067‐1504)
Procedure
1 Set up the following reaction of DNA, 2x KAPA HiFi HotStart ReadyMix, and primers:
Volume
Microbial DNA (5 ng/µl)
2.5 µl
Amplicon PCR Forward Primer 1 µM
5 µl
Amplicon PCR Reverse Primer 1 µM
5 µl
2x KAPA HiFi HotStart ReadyMix
12.5 µl
Total
25 μl
Amplicon PCR
Page 6
2 Seal plate and perform PCR in a thermal cycler using the following program:
• 95°C for 3 minutes
• 25 cycles of:
— 95°C for 30 seconds
— 55°C for 30 seconds
— 72°C for 30 seconds
• 72°C for 5 minutes
• Hold at 4°C
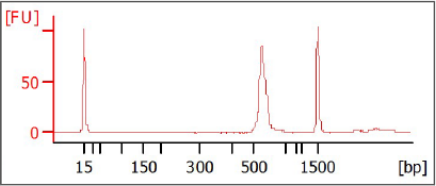
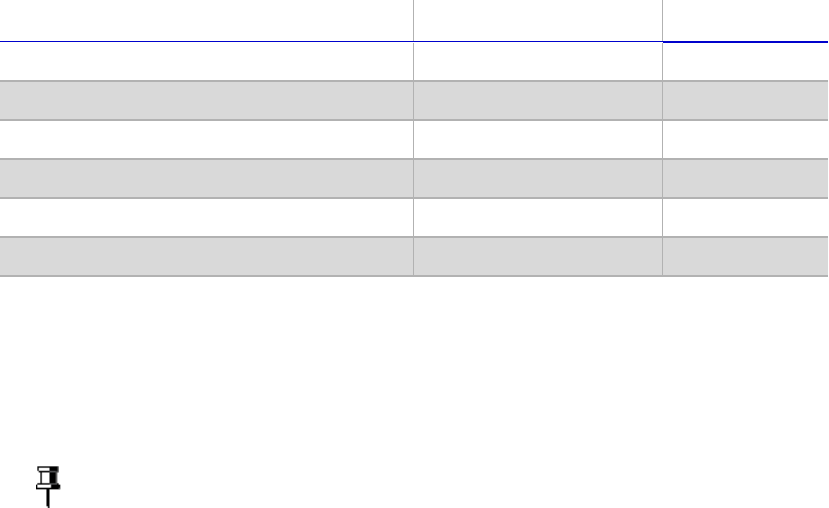
3 [Optional] Run 1 µl of the PCR product on a Bioanalyzer DNA 1000 chip to verify the
size. Using the V3 and V4 primer pairs in the protocol, the expected size on a
Bioanalyzer trace after the Amplicon PCR step is ~550 bp.
Figure 3 Example Bioanalyzer Trace after Amplicon PCR Step
Amplicon PCR
Page 7
PCR Clean‐Up
This step uses AMPure XP beads to purify the 16S V3 and V4 amplicon away from free
primers and primer dimer species.
Consumables
Item Quantity Storage
10 mM Tris pH 8.5 52.5 µl per sample ‐15° to ‐25°C
AMPure XP beads 20 µl per sample 2° to 8°C
Freshly Prepared 80% Ethanol (EtOH) 400 µl per sample
96‐well 0.2 ml PCR plate 1 plate
[Optional] Microseal 'B' film
[Optional] 96‐well MIDI plate 1 plate
Preparation
• Bring the AMPure XP beads to room temperature.
Procedure
1 Centrifuge the Amplicon PCR plate at 1,000 × g at 20°C for 1 minute to collect
condensation, carefully remove seal.
2 [Optional - for use with shaker for mixing] Using a multichannel pipette set to 25 µl,
transfer the entire Amplicon PCR product from the PCR plate to the MIDI plate. Change
tips between samples.
NOTE
Transfer the sample to a 96‐well MIDI plate if planning to use a shaker for mixing. If
mixing by pipette, the sample can remain in the 96‐well PCR plate.
3 Vortex the AMPure XP beads for 30 seconds to make sure that the beads are evenly
dispersed. Add an appropriate volume of beads to a trough depending on the number of
samples processing.
4 Using a multichannel pipette, add 20 µl of AMPure XP beads to each well of the
Amplicon PCR plate. Change tips between columns.
5 Gently pipette entire volume up and down 10 times if using a 96‐well PCR plate or seal
plate and shake at 1800 rpm for 2 minutes if using a MIDI plate.
6 Incubate at room temperature without shaking for 5 minutes.
7 Place the plate on a magnetic stand for 2 minutes or until the supernatant has cleared.
8 With the Amplicon PCR plate on the magnetic stand, use a multichannel pipette to
remove and discard the supernatant. Change tips between samples.
PCR Clean-Up
Page 8

9 With the Amplicon PCR plate on the magnetic stand, wash the beads with freshly
prepared 80% ethanol as follows:
a Using a multichannel pipette, add 200 µl of freshly prepared 80% ethanol to each
sample well.
b Incubate the plate on the magnetic stand for 30 seconds.
c Carefully remove and discard the supernatant.
10 With the Amplicon PCR plate on the magnetic stand, perform a second ethanol wash as
follows:
a Using a multichannel pipette, add 200 µl of freshly prepared 80% ethanol to each
sample well.
b Incubate the plate on the magnetic stand for 30 seconds.
c Carefully remove and discard the supernatant.
d Use a P20 multichannel pipette with fine pipette tips to remove excess ethanol.
11 With the Amplicon PCR plate still on the magnetic stand, allow the beads to air‐dry for
10 minutes.
12 Remove the Amplicon PCR plate from the magnetic stand. Using a multichannel pipette,
add 52.5 µl of 10 mM Tris pH 8.5 to each well of the Amplicon PCR plate.
13 Gently pipette mix up and down 10 times, changing tips after each column (or seal plate
and shake at 1800 rpm for 2 minutes). Make sure that beads are fully resuspended.
14 Incubate at room temperature for 2 minutes.
15 Place the plate on the magnetic stand for 2 minutes or until the supernatant has cleared.
16 Using a multichannel pipette, carefully transfer 50µl of the supernatant from the
Amplicon PCR plate to a new 96‐well PCR plate. Change tips between samples to avoid
cross‐contamination.
SAFE STOPPING POINT
If you do not immediately proceed to Index PCR, seal plate with Microseal “B”
adhesive seal and store it at ‐15° to ‐25°C for up to a week.
PCR Clean-Up
Page 9
Index PCR
This step attaches dual indices and Illumina sequencing adapters using the Nextera XT
Index Kit.
Consumables
Item Quantity Storage
2x KAPA HiFi HotStart ReadyMix
25 µl per sample
‐15° to ‐25°C
Nextera XT Index 1 Primers (N7XX) from the
Nextera XT Index kit
(FC‐131‐1001 or FC‐131‐1002)
5 µl per sample ‐15° to ‐25°C
Nextera XT Index 2 Primers (S5XX) from the
Nextera XT Index kit (FC‐131‐1001 or FC‐131‐
1002)
5 µl per sample ‐15° to ‐25°C
PCR Grade Water 10 µl per sample
TruSeq Index Plate Fixture (FC‐130‐1005) 1
96‐well 0.2 ml PCR plate 1 plate
Microseal 'A' film 1
Procedure
1 Using a multichannel pipette, transfer 5 µl from each well to a new 96‐well plate. The
remaining 45 µl is not used in the protocol and can be stored for other uses.
2 Arrange the Index 1 and 2 primers in a rack (i.e. the TruSeq Index Plate Fixture) using
the following arrangements as needed:
a Arrange Index 2 primer tubes (white caps, clear solution) vertically, aligned with
rows A through H.
b Arrange Index 1 primer tubes (orange caps, yellow solution) horizontally, aligned
with columns 1 through 12.
For more information on index selection, see Dual Indexing Principle, on page 23.
Index PCR
Page 10
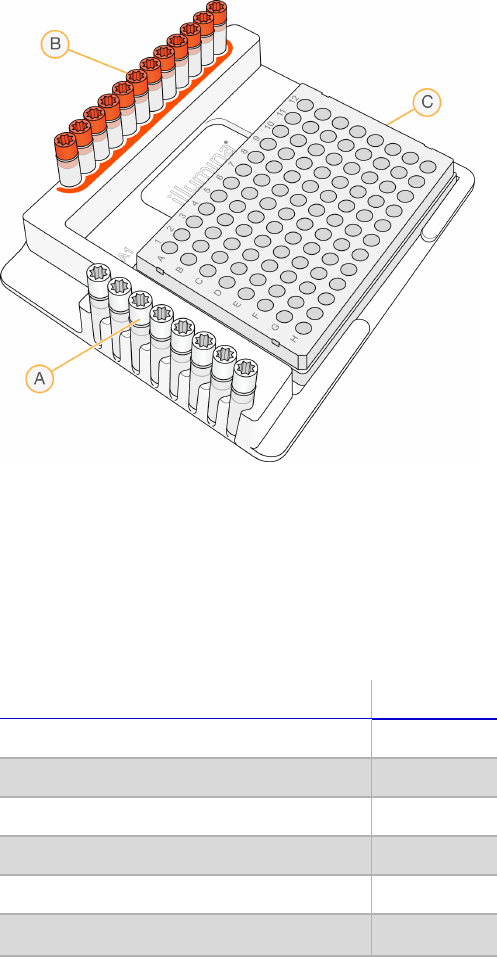
Figure 4 TruSeq Index Plate Fixture
A Index 2 primers (white caps)
B Index 1 primers (orange caps)
C 96‐well plate
3 Place the 96‐well PCR plate with the 5 µl of resuspended PCR product DNA in the
TruSeq Index Plate Fixture.
4 Set up the following reaction of DNA, Index 1 and 2 primers, 2x KAPA HiFi HotStart
ReadyMix, and PCR Grade water:
Volume
DNA
5 µl
Nextera XT Index Primer 1 (N7xx)
5 µl
Nextera XT Index Primer 2 (S5xx)
5 µl
2x KAPA HiFi HotStart ReadyMix
25 µl
PCR Grade water
10 µl
Total
50 μl
5 Gently pipette up and down 10 times to mix.
6 Cover the plate with Microseal 'A'.
7 Centrifuge the plate at 1,000 × g at 20°C for 1 minute.
Index PCR
Page 11
8 Perform PCR on a thermal cycler using the following program:
• 95°C for 3 minutes
• 8 cycles of:
— 95°C for 30 seconds
— 55°C for 30 seconds
— 72°C for 30 seconds
• 72°C for 5 minutes
• Hold at 4°C
Index PCR
Page 12
PCR Clean‐Up 2
This step uses AMPure XP beads to clean up the final library before quantification.
Consumables
Item Quantity Storage
10 mM Tris pH 8.5 27.5 µl per sample ‐15° to ‐25°C
AMPure XP beads 56 µl per sample 2° to 8°C
Freshly Prepared 80% Ethanol (EtOH) 400 µl per sample
96‐well 0.2 ml PCR plate 1 plate
[Optional] Microseal 'B' film
[Optional] 96‐well MIDI plate 1 plate
Procedure
1 Centrifuge the Index PCR plate at 280 × g at 20°C for 1 minute to collect condensation.
2 [Optional - for use with shaker for mixing] Using a multichannel pipette set to 50 µl,
transfer the entire Index PCR product from the PCR plate to the MIDI plate. Change tips
between samples.
NOTE
Transfer the sample to a 96‐well MIDI plate if planning to use a shaker for mixing. If
mixing by pipette, the sample can remain in the 96‐well PCR plate.
3 Vortex the AMPure XP beads for 30 seconds to make sure that the beads are evenly
dispersed. Add an appropriate volume of beads to a trough.
4 Using a multichannel pipette, add 56 µl of AMPure XP beads to each well of the Index
PCR plate.
5 Gently pipette mix up and down 10 times if using a 96‐well PCR plate or seal plate and
shake at 1800 rpm for 2 minutes if using a MIDI plate.
6 Incubate at room temperature without shaking for 5 minutes.
7 Place the plate on a magnetic stand for 2 minutes or until the supernatant has cleared.
8 With the Index PCR plate on the magnetic stand, use a multichannel pipette to remove
and discard the supernatant. Change tips between samples.
9 With the Index PCR plate on the magnetic stand, wash the beads with freshly prepared
80% ethanol as follows:
a Using a multichannel pipette, add 200 µl of freshly prepared 80% ethanol to each
sample well.
b Incubate the plate on the magnetic stand for 30 seconds.
c Carefully remove and discard the supernatant.
PCR Clean-Up 2
Page 13

10 With the Index PCR plate on the magnetic stand, perform a second ethanol wash as
follows:
a Using a multichannel pipette, add 200 µl of freshly prepared 80% ethanol to each
sample well.
b Incubate the plate on the magnetic stand for 30 seconds.
c Carefully remove and discard the supernatant.
d Use a P20 multichannel pipette with fine pipette tips to remove excess ethanol.
11 With the Index PCR plate still on the magnetic stand, allow the beads to air‐dry for 10
minutes.
12 Remove the Index PCR plate from the magnetic stand. Using a multichannel pipette,
add 27.5 µl of 10 mM Tris pH 8.5 to each well of the Index PCR plate.
13 If using a 96‐well PCR plate, gently pipette mix up and down 10 times until beads are
fully resuspended, changing tips after each column. If using a MIDIplate, seal plate and
shake at 1800 rpm for 2 minutes.
14 Incubate at room temperature for 2 minutes.
15 Place the plate on the magnetic stand for 2 minutes or until the supernatant has cleared.
16 Using a multichannel pipette, carefully transfer 25µl of the supernatant from the Index
PCR plate to a new 96‐well PCR plate. Change tips between samples to avoid cross‐
contamination.
SAFE STOPPING POINT
If you do not plan to proceed to Library Quantification, Normalization, and Pooling, on page
16, seal the plate with Microseal “B” adhesive seal. Store the plate at ‐15° to ‐25°C for
up to a week.
PCR Clean-Up 2
Page 14
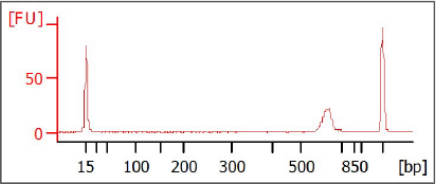
[Optional] Validate Library
Run 1 µl of a 1:50 dilution of the final library on a Bioanalyzer DNA 1000 chip to verify the
size. Using the V3 and V4 primer pairs in the protocol, the expected size on a Bioanalyzer
trace of the final library is ~630 bp.
Figure 5 Example Bioanalyzer Trace of Final Library
[Optional] Validate Library
Page 15

Library Quantification, Normalization, and Pooling
Illumina recommends quantifying your libraries using a fluorometric quantification method
that uses dsDNA binding dyes.
Calculate DNA concentration in nM, based on the size of DNA amplicons as determined by
an Agilent Technologies 2100 Bioanalyzer trace:
(concentration in ng/µl)
(660 g/mol × average library size)
× 10
6
= concentration in nM
For example:
15 ng/µl
(660 g/mol × 500)
× 10
6
= 45 nM
Dilute concentrated final library using Resuspension Buffer (RSB) or 10 mM Tris pH 8.5 to 4
nM. Aliquot 5 µl of diluted DNA from each library and mix aliquots for pooling libraries
with unique indices. Depending on coverage needs, up to 96 libraries can be pooled for one
MiSeq run.
For metagenomics samples, >100,000 reads per sample is sufficient to fully survey the
bacterial composition. This number of reads allows for sample pooling to the maximum
level of 96 libraries, given the MiSeq output of > 20 million reads.
Library Quantification, Normalization, and Pooling
Page 16
Library Denaturing and MiSeq Sample Loading
In preparation for cluster generation and sequencing, pooled libraries are denatured with
NaOH, diluted with hybridization buffer, and then heat denatured before MiSeq sequencing.
Each run must include a minimum of 5% PhiX to serve as an internal control for these low‐
diversity libraries. Illumina recommends using MiSeq v3 reagent kits for improved run
metrics.
Consumables
Item Quantity Storage
10 mM Tris pH 8.5 or RSB (Resuspension Buffer) 6 µl ‐15° to ‐25°C
HT1 (Hybridization Buffer) 1540 µl ‐15° to ‐25°C
0.2 N NaOH (less than a week old) 10 µl ‐15° to ‐25°C
PhiX Control Kit v3 (FC‐110‐3001) 4 µl ‐15° to ‐25°C
MiSeq reagent cartridge 1 cartridge ‐15° to ‐25°C
1.7 ml microcentrifuge tubes (screw cap
recommended)
3 tubes
2.5 L ice bucket
Preparation
1 Set a heat block suitable for 1.7 ml microcentrifuge tubes to 96°C
2 Remove a MiSeq reagent cartridge from ‐15°C to ‐25°C storage and thaw at room
temperature.
3 In an ice bucket, prepare an ice‐water bath by combining 3 parts ice and 1 part water.
Denature DNA
1 Combine the following volumes of pooled final DNA library and freshly diluted 0.2 N
NaOH in a microcentrifuge tube:
• 4 nM pooled library (5 µl)
• 0.2 N NaOH (5 µl)
2 Set aside the remaining dilution of 0.2 N NaOH to prepare a PhiX control within the
next 12 hours.
3 Vortex briefly to mix the sample solution, and then centrifuge the sample solution at 280
× g at 20°C for 1 minute.
4 Incubate for 5 minutes at room temperature to denature the DNA into single strands.
5 Add the following volume of pre‐chilled HT1 to the tube containing denatured DNA:
• Denatured DNA (10 µl)
Library Denaturing and MiSeq Sample Loading
Page 17

• Pre‐chilled HT1 (990 µl)
Adding the HT1 results in a 20 pM denatured library in 1 mM NaOH.
6 Place the denatured DNA on ice until you are ready to proceed to final dilution.
Dilute Denatured DNA
1 Dilute the denatured DNA to the desired concentration using the following example:
NOTE
Illumina recommends targeting 800–1000 K/mm² raw cluster densities using MiSeq v3
reagents. It is suggested to start your first run using a 4 pM loading concentration and
adjust subsequent runs appropriately.
Final
Concentration
2 pM 4 pM 6 pM 8 pM 10 pM
20 pM denatured
library
60 µl 120 µl 180 µl 240 µl 300 µl
Pre‐chilled HT1
540 µl 480 µl 420 µl 360 µl 300 µl
2 Invert several times to mix and then pulse centrifuge the DNA solution.
3 Place the denatured and diluted DNA on ice.
Denature and Dilution of PhiX Control
Use the following instructions to denature and dilute the 10 nM PhiX library to the same
loading concentration as the Amplicon library. The final library mixture must contain at
least 5% PhiX.
1 Combine the following volumes to dilute the PhiX library to 4 nM:
• 10 nM PhiX library (2 µl)
• 10 mM Tris pH 8.5 (3 µl)
2 Combine the following volumes of 4 nM PhiX and 0.2 N NaOH in a microcentrifuge
tube:
• 4 nM PhiX library (5 µl)
• 0.2 N NaOH (5 µl)
3 Vortex briefly to mix the 2 nM PhiX library solution.
4 Incubate for 5 minutes at room temperature to denature the PhiX library into single
strands.
5 Add the following volumes of pre‐chilled HT1 to the tube containing denatured PhiX
library to result in a 20 pM PhiX library:
• Denatured PhiX library (10 µl)
• Pre‐chilled HT1 (990 µl)
6 Dilute the denatured 20 pM PhiX library to the same loading concentration as the
Amplicon library as follows:
Library Denaturing and MiSeq Sample Loading
Page 18
Final
Concentration
2 pM 4 pM 6 pM 8 pM 10 pM
20 pM denatured
library
60 µl 120 µl 180 µl 240 µl 300 µl
Pre‐chilled HT1
540 µl 480 µl 420 µl 360 µl 300 µl
7 Invert several times to mix and then pulse centrifuge the DNA solution.
8 Place the denatured and diluted PhiX on ice.
Combine Amplicon Library and PhiX Control
NOTE
The recommended PhiX control spike‐in of ≥ 5% for low diversity libraries is possible with
RTA v1.17.28 or later, which is bundled with MCS v2.2. For optimal performance, update to v3
software (MCS 2.3). If you are using an older version of the MiSeq software or sequencing
these libraries on the GA or HiSeq, Illumina recommends using ≥ 25% PhiX control spike‐in.
1 Combine the following volumes of denatured PhiX control library and your denatured
amplicon library in a microcentrifuge tube:
• Denatured and diluted PhiX control (30 µl)
• Denatured and diluted amplicon library (570 µl)
2 Set the combined sample library and PhiX control aside on ice until you are ready to
heat denature the mixture immediately before loading it onto the MiSeq v3 reagent
cartridge.
3 Using a heat block, incubate the combined library and PhiX control tube at 96°C for 2
minutes.
4 After the incubation, invert the tube 1–2 times to mix and immediately place in the ice‐
water bath.
5 Keep the tube in the ice‐water bath for 5 minutes.
NOTE
Perform the heat denaturation step immediately before loading the library into the MiSeq
reagent cartridge to ensure efficient template loading on the MiSeq flow cell.
Library Denaturing and MiSeq Sample Loading
Page 19
MiSeq Reporter Metagenomics Workflow
After samples are loaded, the MiSeq system provides on‐instrument secondary analysis
using the MiSeq Reporter software (MSR). MSR provides several options for analyzing
MiSeq sequencing data. For this demonstrated 16S protocol, select the Metagenomics
workflow.
By following this 16S Metagenomics protocol, the Metagenomics workflow classifies
organisms from your V3 and V4 amplicon using a database of 16S rRNA data. The
classification is based on the Greengenes database (http://greengenes.lbl.gov/). The output of
this workflow is a classification of reads at several taxonomic levels: kingdom, phylum,
class, order, family, genus, and species. The analysis output includes:
• Clusters Graph – shows numbers of raw cluster, clusters passing filter, clusters that did
not align, clusters not associated with an index, and duplicates.
• Sample Table – summarizes the sequencing results for each sample.
• Cluster Pie Chart – a graphical representation of the classification breakdown for each
sample.
See the MiSeq Reporter Metagenomics Workflow – Reference Guide (Part # 15042317) for detailed
instructions and guidance.
The method described in this 16S Metagenomics protocol can be used for any targeted
amplicon sequencing, relevant to virus research, mutation detection, or other microbiology‐
related studies. If you use the protocol for other targeted amplicon sequencing studies, select
the MiSeq Reporter Generate FASTQ Workflow for on‐instrument generation of FASTQ files
for downstream analysis. For specific guidance on the Generate FASTQ Workflow, see the
MiSeq Reporter Generate FASTQ Workflow – Reference Guide (Part # 15042322).
MiSeq Reporter Metagenomics Workflow
Page 20

Supporting Information
The protocols described in this guide assume that you are familiar with the contents of this
section and have obtained all of the requisite equipment and consumables.
Acronyms
Acronym
Definition
HT1
Hybridization Buffer
IEM
Illumina Experiment Manager
MSR
MiSeq Reporter
PCR
Polymerase Chain Reaction
rRNA
Ribosomal RNA
RSB
Resuspension Buffer
Table 1 Acronyms
Consumables and Equipment
Check to make sure that you have all of the necessary user‐supplied consumables and
equipment before proceeding to sample preparation.
Consumable Supplier
1.7 ml microcentrifuge tubes General lab supplier
10 µl barrier pipette tips General lab supplier
10 µl multichannel pipettes General lab supplier
10 µl single channel pipettes General lab supplier
20 µl barrier pipette tips General lab supplier
20 µl multichannel pipettes General lab supplier
20 µl single channel pipettes General lab supplier
200 µl barrier pipette tips General lab supplier
200 µl multichannel pipettes General lab supplier
200 µl single channel pipettes General lab supplier
1000 µl barrier pipette tips General lab supplier
Table 2 User‐Supplied Consumables
Supporting Information
Page 21
Consumable Supplier
1000 µl multichannel pipettes General lab supplier
1000 µl single channel pipettes General lab supplier
96‐well 0.2 ml skirtless PCR plates
or
Twin.Tec 96‐well PCR plates
Bio‐Rad, part # MSP‐9601
Agencourt AMPure XP 60 ml kit Beckman Coulter Genomics,
part#A63881
Ethanol 200proof (absolute) for molecular biology
(500ml)
Sigma‐Aldrich, part# E7023
Amplicon PCR Forward Primer (Standard desalting)
Amplicon PCR Reverse Primer (Standard desalting)
KAPA HiFi HotStart ReadyMix (2X) KAPA Biosystems, part # KK2601
Microseal ‘A’ adhesive seals Bio‐Rad, part#MSA‐5001
Microseal ‘B’ adhesive seals Bio‐Rad, part#MSB‐1001
MiSeq Reagent Kit v3 (600 cycle) Illumina, catalog #MS‐102‐3003
Nextera XT Index Kit Illumina, catalog #FC‐131‐1001
or
Illumina, catalog # FC‐131‐1002
PhiX Control Kit v3 Illumina, catalog # FC‐110‐3001
PCR grade water General lab supplier
Fluorometric quantitation with dsDNA binding dye
reagents
General lab supplier
RNase/DNase‐free 8‐well PCR strip tubes and caps General lab supplier
RNase/DNase‐free multichannel reagent reservoirs,
disposable
VWR, part # 89094‐658
Tris‐HCl 10 mM, pH 8.5 General lab supplier
[Optional] 96‐well storage plates, round well, 0.8 ml
(“MIDI” plate)
Fisher Scientific, part#AB‐0859
Equipment Supplier
2.5 L ice bucket General lab supplier
96‐well thermal cycler
(with heated lid)
General lab supplier
Table 3 User‐Supplied Equipment
Supporting Information
Page 22
Equipment Supplier
Fluorometer for quantitation with
dsDNA binding dyes
General lab supplier
Magnetic stand‐96 Life Technologies, catalog#AM10027
Microplate centrifuge General lab supplier
TruSeq Index Plate Fixture Kit (reusable) Illumina, catalog #FC‐130‐1005
[Optional] 2100 Bioanalyzer Desktop System Agilent, part#G2940CA
[Optional] Agilent DNA 1000 Kit Agilent, part#5067‐1504
[Optional] High Speed Micro Plate Shaker VWR, catalog # 13500‐890 (110V/120V)
or
VWR, catalog # 14216‐214 (230V)
Dual Indexing Principle
The dual indexing strategy uses two 8 base indices, Index 1 (i7) adjacent to the P7 sequence,
and Index 2 (i5) adjacent to the P5 sequence. Dual indexing is enabled by adding a unique
Index 1 (i7) and Index 2 (i5) to each sample. The 96 sample Nextera XT Index Kit (FC‐131–
1002) use 12 different Index 1 (i7) adapters (N701–N712) and 8 different Index 2 (i5)
adapters (S501–S508). The 24 sample Nextera XT Index Kit (FC‐131–1001) uses 6 different
Index 1 (i7) adapters (N701–N706) and 4 different Index 2 (i5) adapters (S501–S504). In the
Index adapter name, the N or S refers to Nextera XT sample preparation, 7 or 5 refers to
Index 1 (i7) or Index 2 (i5), respectively. The 01–12 refers to the Index number. A list of index
sequences is provided for generating sample sheets to demultiplex the samples:
Index 1 (i7) Sequence Index 2 (i5) Sequence
N701 TAAGGCGA S501 TAGATCGC
N702 CGTACTAG S502 CTCTCTAT
N703 AGGCAGAA S503 TATCCTCT
N704 TCCTGAGC S504 AGAGTAGA
N705 GGACTCCT S505 GTAAGGAG
N706 TAGGCATG S506 ACTGCATA
N707 CTCTCTAC S507 AAGGAGTA
N708 CAGAGAGG S508 CTAAGCCT
N709 GCTACGCT
N710 CGAGGCTG
N711 AAGAGGCA
N712 GTAGAGGA
Low Plexity Pooling Guidelines
Illumina uses a green laser or LED to sequence G/T and a red laser or LED to sequence A/C.
At each cycle, at least one of two nucleotides for each color channel are read to ensure proper
registration. It is important to maintain color balance for each base of the index read being
sequenced, otherwise index read sequencing could fail due to registration failure. If you
choose the dual‐indexed sequencing workflow, always use at least two unique and
Supporting Information
Page 23
compatible barcodes for each index (index 1 and index 2). The following tables illustrate
possible pooling strategies:
Plex Index 1 (i7) Selection Index 2 (i5) Selection
1‐plex (no
pooling)
Any Index 1 adapter Any Index 2 adapter
2‐plex • [option 1] N702 and N701
• [option 2] N702 and N704
3‐plex • [option 1] N701, N702, and N704
• [option 2] N703, N705, and N706
4‐ or 5‐plex • [option 1] N701, N702, N704, and
any other Index 1 adapter
• [option 2] N703, N705, N706, and
any other Index 1 adapter
6‐plex N701, N702, N703, N704, N705, and
N706
Table 4 Libraries Pooled: 6 or fewer; Sequencing Workflow: Single Index
Plex Index 1 (i7) Selection Index 2 (i5) Selection
7–12 plex, Dual
Index
• [option 1] N701, N702, N704, and
any other Index 1 adapter (as
needed)
• [option 2] N703, N705, N706, and
any other Index 1 adapter (as
needed)
• [option 1] S501 and S502
• [option 2] S503 and S504
• [option 3] S505 and S506
7–12 plex, Single
Index
(96 sample
Nextera Index
adapter kit)
• N701–N706 and any other Index 1
adapter (as needed)
• Any Index 2 (i5) adapter
Greater than 12‐
plex
N701, N702, N703, N704, N705,
N706, and any other Index 1 adapter
• [option 1] S501, S502, and any
other Index 2 adapter (as needed)
• [option 2] S503, S504, and any
other Index 2 adapter (as needed)
• [option 3] S505, S506, and any
other Index 2 adapter (as needed)
Table 5 Sequencing Workflow: Single or Dual Index
Supporting Information
Page 24
These strategies represent only some of the acceptable combinations. Alternatively, check the
real sequences of each index in the tables to make sure that each base position has a signal
in both color channels for the index read:
Good Bad
Index 1 Index 2 Index 1 Index 2
705 GGACTCCT 503 TATCCTCT 705 GGACTCCT 502 CTCTCTAT
706 TAGGCATG 503 TATCCTCT 706 TAGGCATG 502 CTCTCTAT
701 TAAGGCGA 504 AGAGTAGA 701 TAAGGCGA 503 TATCCTCT
702 CGTACTAG 504 AGAGTAGA 702 CGTACTAG 503 TATCCTCT
√ √ √ √ √ √ √ √ √ √ √ √ √ √ √ √
√ √ √ √ √ √ √ √
√ √ √ √ xxxx
√=signal in both color
x=signal missing in one color channel
Prevent PCR Product Contamination
The PCR process is commonly used in the laboratory to amplify specific DNA sequences.
Unless proper laboratory hygiene is used, PCR products can contaminate reagents,
instrumentation, and genomic DNA samples, causing inaccurate and unreliable results. PCR
product contamination can shut down lab processes and significantly delay normal
operations.
Make sure that the lab is set up appropriately to reduce the risk of PCR product
contamination:
• Physically Separate Pre-PCR and Post-PCR Areas
• Physically separate laboratory space where pre‐PCR processes are performed (DNA
extraction, quantification, and normalization) from the laboratory space where PCR
products are made and processed (post‐PCR processes).
• Never use the same sink to wash pre‐PCR and post‐PCR troughs.
• Never share water purification systems for pre‐PCR and post‐PCR processes.
• Store all supplies used in the protocols in the pre‐PCR area, and transfer to the post‐
PCR area as needed.
• Use Dedicated Equipment and Supplies
• Dedicate separate full sets of equipment and supplies (pipettes, centrifuges, oven,
heat block, etc.) to pre‐PCR and post‐PCR lab processes, and never share between
processes.
• Dedicate separate storage areas (freezers and refrigerators) to pre‐PCR and post‐PCR
consumables.
Because the pre‐ and post‐amplification reagents are shipped together, it is important to
unpack the reagents in the pre‐PCR lab area. After unpacking the reagents, move the post‐
amplification reagents to the proper post‐PCR storage area.
Pre‐PCR and Post‐PCR Lab Procedures
To prevent PCR product contamination, it is important to establish lab procedures and
follow best practices. Illumina recommends daily and weekly cleaning of lab areas using
Supporting Information
Page 25
0.5% Sodium Hypochlorite (10% Bleach).
CAUTION
To prevent sample or reagent degradation, make sure that all vapors from the cleaning
solution have fully dissipated before beginning any processes.
Daily Cleaning of Pre‐PCR Area
A daily cleaning of the pre‐PCR area using a 0.5% Sodium Hypochlorite (10% Bleach)
solution helps to eliminate PCR product that has entered the pre‐PCR area.
Identify pre‐PCR areas that pose the highest risk of contamination, and clean these areas
with a 0.5% Sodium Hypochlorite (10% Bleach) solution before beginning any pre‐PCR
processes. High‐risk areas might include, but are not limited to, the following items:
• Benchtops
• Door handles
• Refrigerator/freezer door handles
• Computer mouse
• Keyboards
Daily Cleaning of Post‐PCR Area
Reducing the amount of PCR product in the post‐PCR area helps reduce the risk of
contamination in the pre‐PCR area. Daily cleaning of the post‐PCR area using a 0.5%
Sodium Hypochlorite (10% Bleach) solution helps reduce the risk of contamination.
Identify post‐PCR areas that pose the highest risk of contamination, and clean these areas
with a 0.5% Sodium Hypochlorite (10% Bleach) solution daily. High‐risk areas might
include, but are not limited to, the following items:
• Thermal cyclers
• Bench space used to process amplified DNA
• Door handles
• Refrigerator/freezer door handles
• Computer mouse
• Keyboards
Weekly Cleaning of All Lab Areas
One time a week, perform a thorough cleaning of the pre‐PCR and post‐PCR areas using
0.5% Sodium Hypochlorite (10% Bleach).
• Clean all benchtops and laboratory surfaces.
• Clean all instruments that are not cleaned daily.
• Thoroughly mop lab floors.
• Make sure that personnel responsible for weekly cleaning are properly trained on
prevention of PCR product contamination.
Items Fallen to the Floor
The floor is contaminated with PCR product transferred on the shoes of individuals coming
from the post‐PCR area; therefore, anything falling to the floor must be treated as
contaminated.
• Disposable items that have fallen to the floor, such as empty tubes, pipette tips, gloves,
lab coat hangers, must be discarded.
Supporting Information
Page 26
• Non‐disposable items that have fallen to the floor, such as a pipette or an important
sample container, must be immediately and thoroughly cleaned. Use a 0.5% Sodium
Hypochlorite (10% Bleach) solution to remove PCR product contamination.
• Clean any lab surface that has come in contact with the contaminated item. Individuals
handling anything that has fallen to the floor, disposable or non‐disposable, must
discard their lab gloves and put on a new pair.
Best Practices
When preparing libraries for sequencing, always adhere to good molecular biology practices.
Read through the entire protocol before starting to make sure that all of the required
materials are available and your equipment is programmed and ready to use.
Handling Liquids
Good liquid handling measures are essential, particularly when quantifying libraries or
diluting concentrated libraries for making clusters.
• Small differences in volumes (±0.5µl) can sometimes cause large differences in cluster
numbers (~100,000).
• Small volume pipetting can be a source of potential error in protocols requiring the
generation of standard curves, such as qPCR, or small but precise volumes, such as the
Agilent Bioanalyzer.
• If small volumes are unavoidable, use due diligence to make sure that pipettes are
correctly calibrated.
• Make sure that pipettes are not used at the volume extremes of their performance
specifications.
• Prepare the reagents for multiple samples simultaneously, to minimize pipetting errors,
especially with small volume enzyme additions. As a result, pipette one time from the
reagent tubes with a larger volume, rather than many times with small volumes. Aliquot
to individual samples in a single pipetting movement to allow for standardization
across multiple samples.
Handling Magnetic Beads
NOTE
Cleanup procedures have only been validated using the 96‐well plates and the magnetic stand
specified in the Consumables and Equipment list. Comparable performance is not guaranteed
when using a microcentrifuge tube or other formats, or other magnets.
• Before use, allow the beads to come to room temperature.
• Do not reuse beads. Always add fresh beads when performing these procedures.
• Immediately before use, vortex the beads until they are well dispersed and the color of
the liquid is homogeneous.
• When pipetting the beads, pipette slowly and dispense slowly due to the viscosity of the
solution.
• Take care to minimize bead loss, which can affect final yields.
• Change the tips for each sample, unless specified otherwise.
• Let the mixed samples incubate at room temperature for the time indicated in the
protocol for maximum recovery.
Supporting Information
Page 27
• When removing and discarding supernatant from the wells, use a single channel or
multichannel pipette and take care not to disturb the beads.
• When aspirating the cleared solution from the reaction plate and wash step, it is
important to keep the plate on the magnetic stand and not disturb the separated
magnetic beads. Aspirate slowly to prevent the beads from sliding down the sides of the
wells and into the pipette tips.
• To prevent the carryover of beads after elution, approximately 2.5 µl of supernatant is
left when the eluates are removed from the bead pellet.
• Be sure to remove all of the ethanol from the bottom of the wells, as it can contain
residual contaminants.
• Keep the reaction plate on the magnetic stand and let it air‐dry at room temperature to
prevent potential bead loss due to electrostatic forces. Allow for the complete
evaporation of residual ethanol, because the presence of ethanol affects the performance
of the subsequent reactions. Illumina recommends at least minutes drying time, but a
longer drying time can be required. Remaining ethanol can be removed with a 10µl
pipette.
• Avoid over drying the beads, which can impact final yields.
• Do not scrape the beads from the edge of the well using the pipette tip.
• To maximize sample recovery during elution, incubate the sample/bead mix for
2minutes at room temperature before placing the samples onto the magnet.
Avoiding Cross‐Contamination
Practice the following to avoid cross‐contamination:
• Open only one adapter tube at a time.
• Change the tips for each sample, unless specified otherwise.
• Pipette carefully to avoid spillage.
• Clean pipettes and change gloves between handling different adapter stocks.
• Clean work surfaces thoroughly before and after the procedure.
Potential DNA Contaminants
When handling and processing samples using this protocol, use best practices to avoid PCR
contamination, as you would when preparing PCR amplicons.
Temperature Considerations
Temperature is an important consideration for making libraries:
• Keep libraries at temperatures ≤37°C, except where specifically noted.
• Place reagents on ice after thawing at room temperature.
Equipment
• Review the programming instructions for your thermal cycler user guide to make sure
that it is programmed appropriately using the heated lid function.
• It is acceptable to use the thermal cycler tracked heating lid function.
Supporting Information
Page 28
