
March 2023 Issue Brief 1
FDA User Fees: Examining Changes in Medical Product
Development and Economic Benefits
KEY FINDINGS
• The Food and Drug Administration (FDA) assesses and collects user fees to supplement Congressional
appropriations. In fiscal year (FY) 2022, user fees represented 46% ($2.9 billion) of FDA’s total budget of $6.2
billion.
• User fees vary substantially across FDA programs. In FY2022, across FDA’s medical product centers, user fees
accounted for $1.4 billion (66%) of the human drugs program budget, $197 million (43%) of the biologics
program budget, and $228 million (35%) of the medical device program budget.
• Research evidence suggests that user fees have contributed to increased access to new medical products for
patients and reduced review timelines for industry. The benefits of user fees can be substantial for industry
and patients.
• Using public and proprietary data from 2000 to 2018, we estimate that between 0.5% and 2.0% of the total
cost of developing a new drug, complex medical device, or preventive vaccine went towards user fees. Using
data from 2013 to 2021, we estimate that 1.7% of the total cost to develop a generic drug went towards user
fees.
Introduction
The Food and Drug Administration (FDA) relies on funding from two sources to support its regulatory
activities: user fees and budget appropriations and user fees paid by industry. Both sources of funding are
authorized by Congress. FDA collects user fees from the regulated industry; for medical products, this includes
biopharmaceutical and medical device companies. Budget appropriations are borne by the public more broadly
through taxes, while user fees are directly supported by sponsors of medical product applications through
reduced profit margins and indirectly borne by consumers to the extent that fees are passed through to
consumers through increased prices. User fee programs are intended to supplement Congressional
appropriations.
User fees for all human medical products, which includes prescription drugs, medical devices, generic
drugs, and biosimilars, are reauthorized together on a five-year cycle. The reauthorization process begins with
FDA and industry negotiating performance goals, such as completing reviews within a specified timeframe. As
part of the reauthorization process, other relevant stakeholders also provide input on the draft agreement
through public comment. FDA then transmits the final agreement to Congress for approval. On September 30,
2022, the President signed legislation reauthorizing the medical product user fees for an additional five years,
through September 30, 2027.
The goal of this Issue Brief is to provide a primer on FDA user fees, present findings that examine how
user fees affect the costs of medical product development, and summarize the research literature on user fees,
most notably in expediting medical product development and approval.
OFFICE OF
SCIENCE & DATA POLICY
ISSUE BRIEF
MARCH 2023

March 2023 Issue Brief 2
History of User Fees
In the late 1980s, long FDA review times (on average 29 months) for human drug applications raised
concerns from manufacturers that lengthy review times meant lost sales and from patient advocates about
delayed access to new treatments.
1
This was particularly salient in part due to a backlog of applications for new
antibiotics, drugs for heart failure, and therapeutics for the AIDS crisis.
2
Lengthy review times were primarily due
to limited budgetary resources.
Another challenge was that sponsors lacked information about the status of their applications and
predictability around their application review times; industry meetings with FDA were often considered a
privilege because there were not enough resources to support even the required meetings.
2
In response,
industry, with support from patient advocates, agreed to pay user fees beginning in 1992 to supplement the FDA
budget in exchange for agreements to reduce review times. Since the 1950s, Federal agencies, including FDA,
Congress, and the Courts, had deliberated about the merits and legality of user fees for federal programs. These
discussions included the Office of Management and Budget’s guidance on user fees issued in 1959, which
specified that if the benefits of a good or service accrues broadly to the public, then the goods or services should
be financed by taxes paid by the public.
3
By the early 1980s a Supreme Court decision clarified aspects of user
fee programs that opened the way for a user fee structure for federal agencies including FDA. The decision led
to the eventual conclusion that as long as user fees were specifically structured with a “public health
component” funded through budget authority and a “industry benefit” component funded through user fees,
they would be legal.
2
By charging the costs of programs or activities to those that benefit the most from them,
user fees can reduce taxpayer burden, while also reducing uncertainty for industry by giving them predictable
and shorter review timelines.
To date, the authorizing legislation for each user fee program has set an amount of fee revenue for the
first year of the program with annual adjustments for subsequent years, specified the types of fees that may be
assessed and collected, and established legal conditions in order for FDA to collect user fees. It also has
committed FDA to certain performance goals, such as completing reviews within a specified timeframe. Some of
the user fee programs require reauthorization to continue while others are permanently authorized.
4
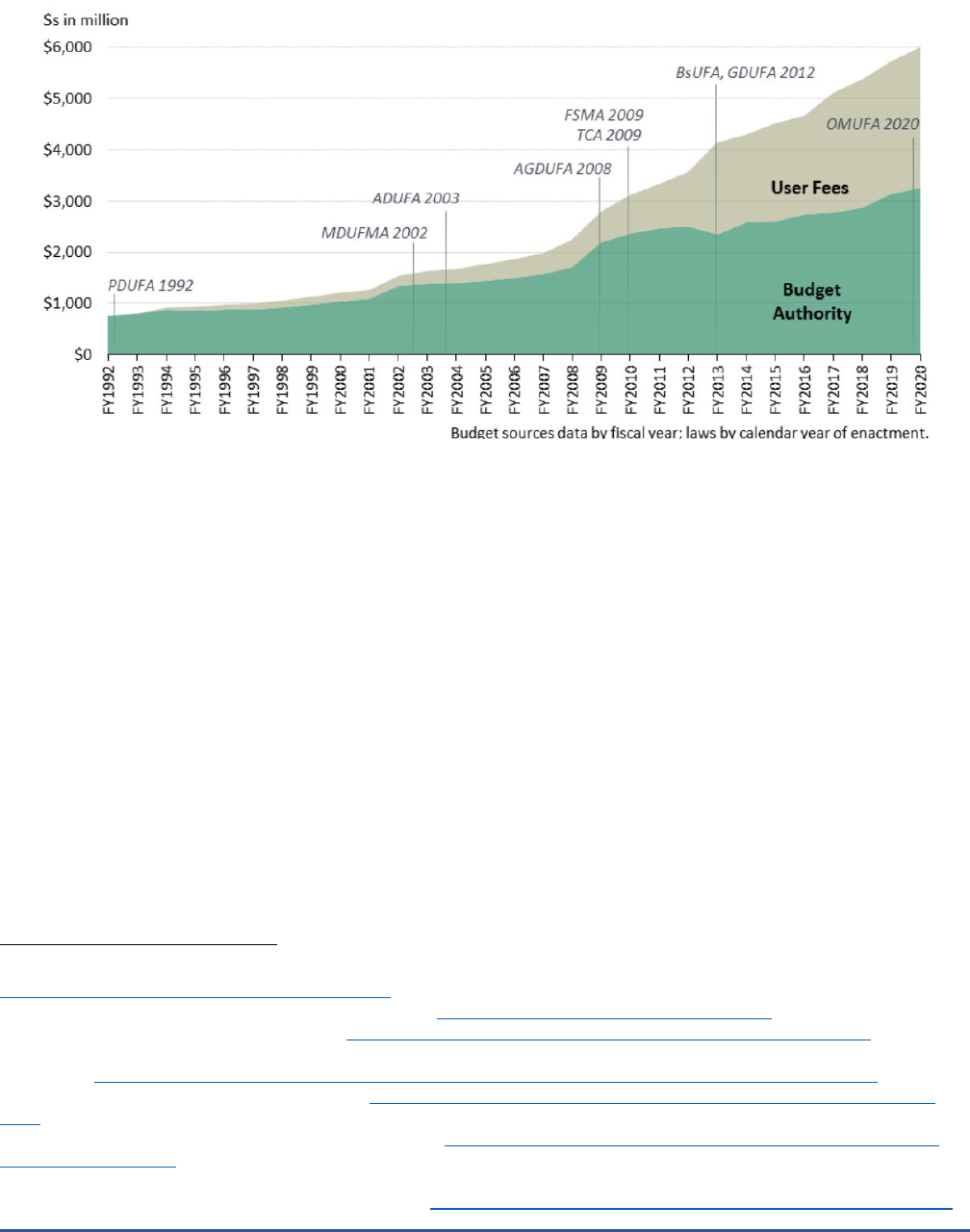
Figure 1 shows FDA’s budget from FY 1992 through FY 2020 and the relative portion of the budget that
is made up of user fees versus budget authority. We see that FDA’s budget has risen over time, with the greatest
increases due to user fees, rather than increases in budget authority. Specifically, FDA’s budget has grown from
just under $1 billion in FY 1992, when it was entirely budget authority, to almost $6 billion in FY 2020,
approximately $3 billion of which was for user fees.
1
Cutler, D.M. (2019). Extending the User Fee Approach to Pharmaceuticals. The JAMA Forum, 320(15): 1525-1526.
2
Woodcock, J & Junod, S. PDUFA Lays the Foundation: Launching into the Era of User Fee Acts U.S. Food and Drug Administration.
https://www.fda.gov/files/about%20fda/published/PDUFA-Lays-the-Foundation--Launching-Into-the-Era-of-User-Fee-Acts.pdf
3
Office of Management and Budget. (1993). Circular No. A-25 Revised. Memorandum for Heads of Executive Departments and
Establishments, Available at: https://obamawhitehouse.archives.gov/omb/circulars_a025/
; last accessed August 28, 2022.
4
For additional details on user fee programs and how they work, see the various Congressional Research Service reports on this topic. For
example: Bodie, A. Sarata, A.K. FDA Human Medical Product User Fee Programs. Congressional Research Service, Report R44750,
September 2021. Available at https://crsreports.congress.gov/product/details?prodcode=R44750.
March 2023 Issue Brief 3
Figure 1. Distribution of User Fees and Budget Authority for FDA, 1992-2020 ($millions)
Source: This graph is reproduced from a report by the Congressional Research Service
5
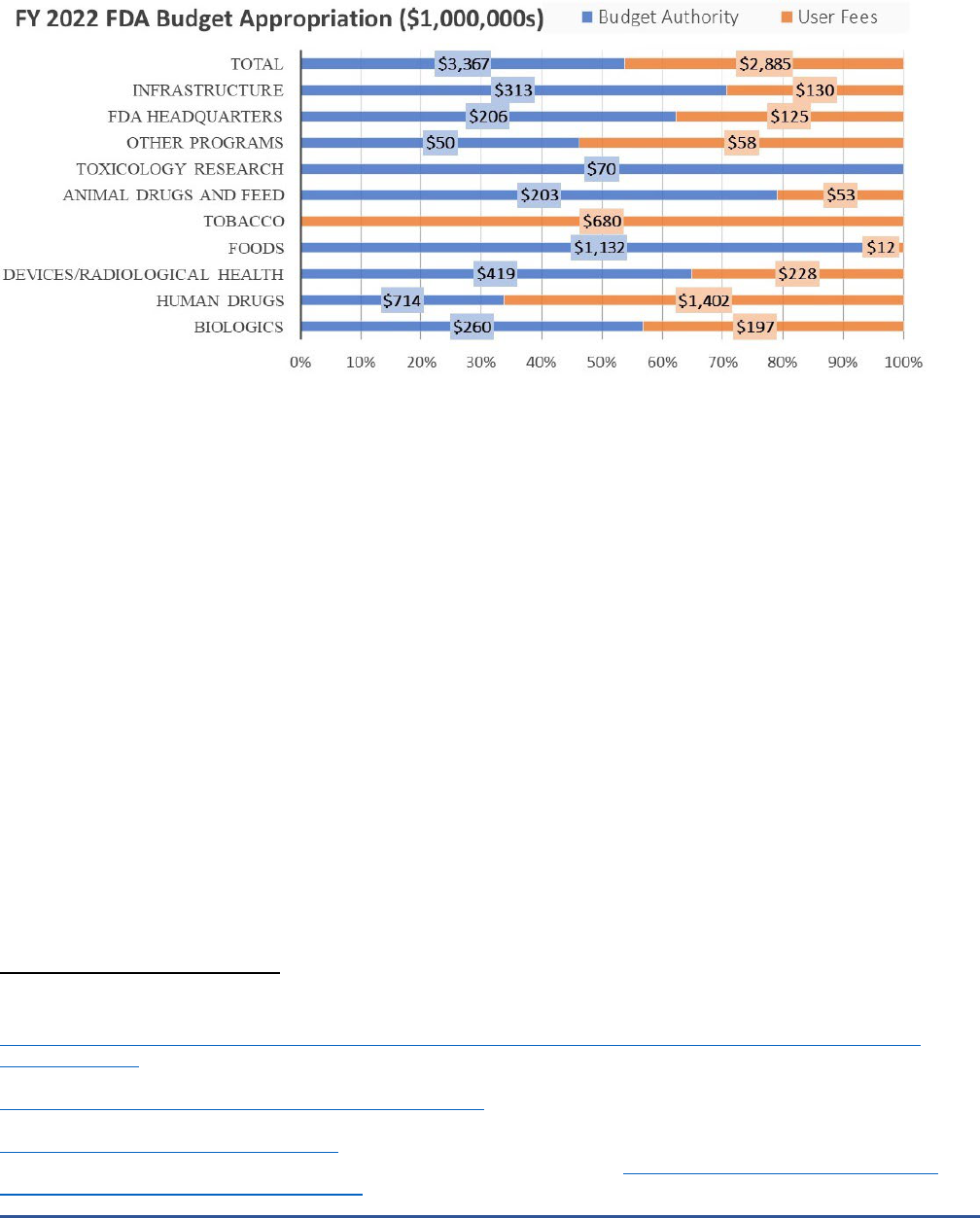
Figure 2 shows how user fees fit into the overall FDA budget. In fiscal year (FY) 2022, across FDA’s
medical product centers, user fees made up approximately 46 percent ($2.9 billion) of FDA’s total operating
budget of $6.2 billion.
6
Focusing specifically on medical products, user fees accounted for $1.4 billion (66
percent) of the human drugs program budget, $197 million (43 percent) of the biologics program budget, and
$228 million (35 percent) of the medical devices program budget.
6
User fees for prescription drugs, including
biologics and generic drugs, and medical devices are the focus of this report; however, additional medical
product user fee programs have also been authorized for biosimilars,
7
over-the-counter drugs,
8
animal drugs,
9
and animal generic drugs.
10
User fees also exist for the other products regulated by FDA, including tobacco
products, which are funded entirely through user fees, and foods, which are funded almost entirely through
budget authority.
11
The money collected through each of the user fee programs has specific allowable and
excluded costs in terms of what FDA is allowed to spend the funds on. Allowable costs are for activities that
directly relate to the respective user fee program (e.g., application review, regulatory research to support the
submission of high-quality applications and reduce time to approval, facility inspections, and post-market safety
surveillance), while excluded costs are all FDA actions that are not specific to the user fee programs, such as
agency infrastructure and other agency wide efforts.
5
Congressional Research Service. (2022). The Food and Drug Administration (FDA) Budget: Fact Sheet. Report # R44576.
https://crsreports.congress.gov/product/pdf/R/R44576
6
U.S. Food and Drug Administration. (2022). FDA At a Glance. https://www.fda.gov/media/154548/download
7
Biosimilar User Fee Act “BsUFA” began in 2012. https://www.fda.gov/industry/fda-user-fee-programs/bsufa-authorization
8
Over -the-Counter Monograph User Fee Act “OMUFA” passed as part of the 2020 Coronavirus Aid, Relief, and Economic Security Act or
“CARES Act”. https://www.fda.gov/industry/fda-user-fee-programs/over-counter-monograph-drug-user-fee-program-omufa
9
Animal Drug User Fee Act “ADUFA” began in 2003. https://www.fda.gov/industry/fda-user-fee-programs/animal-drug-user-fee-act-
adufa
10
Animal Generic Drug User Fee Act “AGDUFA” began in 2008. https://www.fda.gov/industry/fda-user-fee-programs/animal-generic-
drug-user-fee-act-agdufa
11
The 2009 Family Smoking Prevention and Tobacco Control Act authorized FDA to assess and collect user fees that provide 100% of
funding for FDA’s tobacco products and regulatory activities. https://www.fda.gov/tobacco-products/manufacturing/tobacco-user-fees
March 2023 Issue Brief 4
Figure 2. Fiscal Year 2022 FDA Budget Appropriations ($millions)
Prescription Drug User Fee Act
In 1992, the Prescription Drug User Fee Act (PDUFA) was first enacted, authorizing FDA to assess and
collect fees from manufacturers. Since then, a portion of FDA’s budget for the review of drugs and biologics has
come from industry user fees. The original goal of PDUFA was to speed the FDA’s application review process for
new drugs and biological products without compromising FDA’s standards for new drug safety, efficacy, and
quality.
12
Originally, user fees were authorized to support only premarket review activities, which allowed FDA
to hire additional staff to help meet its performance goals. Over time, other activities that relate specifically to
prescription drug development, such as preclinical drug development, certain post-marketing activities, and
enhancements to technology systems, have also been allowed to be paid by user fees.
4,13,14
There are two types of PDUFA fees: application fees and program fees. Application fees are one-time
fees that are required when an application for a new small molecule drug or biological product is submitted to
FDA. The FY2023 application fees are $3,242,026 per application with clinical data and $1,621,013 per
application for which clinical data is not required.
15
Program fees are annual fees paid by all sponsors of
products that are FDA approved and legally marketed. The FY2023 program fees are $393,933 per sponsor.
15
Waivers or adjustments to user fees are permitted for several reasons, including if a waiver or reduction is
necessary to protect the public health, assessment of the user fees would present a significant barrier to
innovation due to limited resources or other circumstances, or the applicant involved is a small business
submitting its first human drug application for review.
12
Testimony of Patrizia Cavazzoni and Peter Marks on February 3, 2022 to the House Committee on Energy and Commerce,
Subcommittee on Health titled “FDA User Fee Reauthorization: Ensuring Safe and Effective Drugs and Biologics”.
https://www.fda.gov/news-events/congressional-testimony/fda-user-fee-reauthorization-ensuring-safe-and-effective-drugs-and-
biologics-02032022
13
Congressional Research Service. (2021). FDA Human Medical Product User Fee Programs (R44750).
https://crsreports.congress.gov/product/details?prodcode=R44750
. Last accessed August 18, 2022.
14
U.S. Food and Drug Administration. (2022). FY2021 Financial Report to Congress for the Prescription Drug User Fee Act of 1992.
https://www.fda.gov/media/156199/download
. Accessed September 14, 2022.
15
U.S. Food and Drug Administration. (2022). Prescription Drug User Fee Amendments. https://www.fda.gov/industry/fda-user-fee-
programs/prescription-drug-user-fee-amendments

March 2023 Issue Brief 5
Medical Device User Fee Amendments
In 2002, Congress enacted the Medical Device User Fee Modernization Act (MDUFMA) to implement a
user fee program for medical devices; MDUFMA is now called the Medical Device User Fee Amendments
(MDUFA). The goal of the legislation was to improve the predictability, transparency, and consistency of
regulatory processes for medical devices to incentivize innovation and get more products to market faster.
16
For
instance, the most recent MDUFA performance goals include agreed-upon timelines for review, process
improvements in the pre-marketing and marketing application processes, improved engagement and
communication with industry, improvements in scientific and regulatory review capacity, enhanced patient
engagement and patient input in the regulatory process, and support for adoption of emerging diagnostics,
regular progress reports, and conduct of an independent assessment of the review of device applications.
17
There are two types of user fees for medical device companies. Device manufacturers pay establishment
registration fees to FDA when they register their establishments, and they pay application fees when they
submit a medical device marketing application for review. User fees are also paid for other submissions, such as
annual reports and 513(g) information requests (a request for the agency’s views about the classification and
the regulatory requirements that may be applicable to a particular device). Generally, establishments that are
required to register are also required to list the devices that are made in each registered establishment.
18
All
establishments must pay the establishment registration fee ($6,493 per establishment for FY2023); there are no
waivers or reductions for small establishments. The application fee differs by the type of submission, ranging
from $5,961 for a 513(g) information request to $441,547 for a premarket approval application (PMA) in FY2023.
Small businesses pay a reduced application fee – the fees range from $2,980 to $110,387—and they can apply
for a waiver for their first marketing application.
19
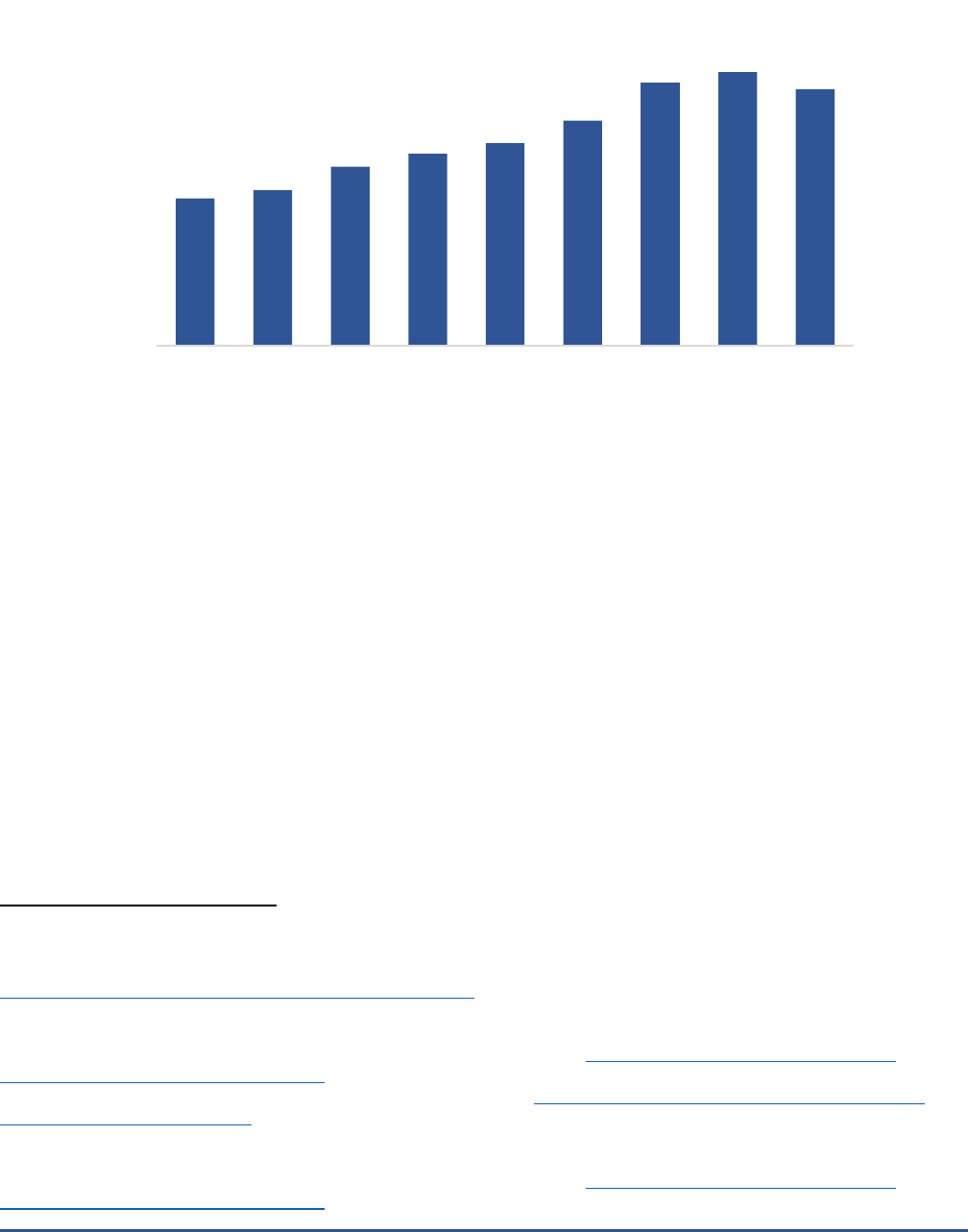
In recent years, FDA has seen unprecedented growth in the number of requests for pre-submission
consultations.
20
MDUFA IV authorized resources for about 2,350 pre-submissions each year, but the number of
pre-submissions over fiscal years 2017-2021 exceeded the anticipated level by up to 1,000 pre-submissions per
year (Figure 3).
16
16
Testimony by Jeffrey Shuren on March 31, 2022 titled “FDA User Fee Reauthorization: Ensuring Safe and Effective Medical Devices”.
https://www.fda.gov/news-events/congressional-testimony/fda-user-fee-reauthorization-ensuring-safe-and-effective-medical-devices-
04012022
17
U.S. Food and Drug Administration. (2023). Medical Device User Fee Amendments 2022 (MDUFA V).
https://www.fda.gov/industry/medical-device-user-fee-amendments-mdufa/medical-device-user-fee-amendments-2022-mdufa-v
18
U.S. Food and Drug Administration. (2021). Device Registration and Listing. https://www.fda.gov/medical-devices/how-study-and-
market-your-device/device-registration-and-listing
19
U.S. Food and Drug Administration. (2022). Medical Device User Fee Amendments (MDUFA). https://www.fda.gov/industry/fda-user-
fee-programs/medical-device-user-fee-amendments-mdufa
20
Pre-submissions follow a structured process for interactions between FDA and industry. These interactions are intended to obtain FDA
feedback on future applications prior to their submission.
March 2023 Issue Brief 6
Figure 3. Requests for Device Pre-Submission Consultations Received by FDA
16,21
Generic Drug User Fee Amendments (GDUFA)
In the years preceding user fees for generic drugs, the number of generic drug applications and the
number of foreign facilities making generic drugs grew to such an extent that FDA experienced a backlog of
marketing applications. This was a result of lack of resources to keep pace with the growth in the industry.
22,23
After negotiation and consultation with stakeholders, FDA and the generic drug industry developed a proposal
for a user fee program for generic drugs and submitted it to Congress. In 2012, the Generic Drug User Fee
Amendments (GDUFA)
24
were passed as part of the Food and Drug Administration Safety and Innovation Act
(FDASIA), which allowed FDA to assess and collect user fees from drug companies that submit marketing
applications for certain generic human drug applications, drug master files holders, and facilities.
GDUFA I (2012 to 2017) enabled FDA to implement enhancements to expedite the review and approval
of human generic drugs. It also brought a risk-based approach to good manufacturing practice inspections with
the goal of achieving parity of inspection frequency between domestic and foreign manufacturers. More recent
goals include reducing the number of review cycles to approval, supporting the development of complex generic
drug products, and improving communications with industry.
25
GDUFA includes four types of fees.
26
Annual
program fees depend on the number of approved generic applications (called abbreviated new drug applications
or ANDAs) per company.
27
In FY2023, program fees ranged from $162,056 to $1,620,556 per company. There
are also facility fees which vary depending on the type of facility (i.e., active pharmaceutical ingredients (API),
21
The definition of a pre-submission changed over time, so it is not possible to have a longer period of observations. Pre-submissions
began to be part of the MDUFA negotiations starting when the data are shown here, hence there is no pre-period data available.
22
Congressional Research Service. (2021). The Generic Drug User Fee Amendments (GDUFA): Background and Reauthorization, R46778.
https://crsreports.congress.gov/product/details?prodcode=R46778
23
Testimony by Janet Woodcock on March 2, 2017 before the House Committee on Energy and Commerce, Subcommittee on Health,
titled “Generic Drug User Fee Act Reauthorization (GDUFA II)”. 115th Cong., 1st session, p. 4.
24
U.S. Food and Drug Administration. (2022). Generic Drug User Fee Amendments. https://www.fda.gov/industry/fda-user-fee-
programs/generic-drug-user-fee-amendments
25
U.S. Food and Drug Administration. (2022). GDUFA Performance Reports. https://www.fda.gov/about-fda/user-fee-performance-
reports/gdufa-performance-reports
26
In addition to the four categories of fees discussed above, GDUFA also includes a one-time backlog fee for abbreviated new drug
applications pending on October 1, 2012 that had not received a tentative approval prior to that date.
27
U.S. Food and Drug Administration. (2022). Generic Drug User Fee Amendments. https://www.fda.gov/industry/fda-user-fee-
programs/generic-drug-user-fee-amendments
1820
1920
2210
2370
2500
2780
3250
3380
3170
0
500
1000
1500
2000
2500
3000
3500
4000
FY2013 FY2014 FY2015 FY2016 FY2017 FY2018 FY2019 FY2020 FY2021
March 2023 Issue Brief 7
finished dosage form and contract manufacturing organizations) and whether a facility is foreign or domestic.
For example, in FY2023 the user fee for an API facility was $37,544 and $52,544 per domestic and foreign
facility, respectively. There are also fees for manufacturers involved in producing certain types of APIs (called
type II active pharmaceutical ingredient drug master file holders) ($78,293 in FY2023) and for filing an ANDA for
FDA review ($240,582 for FY2023).
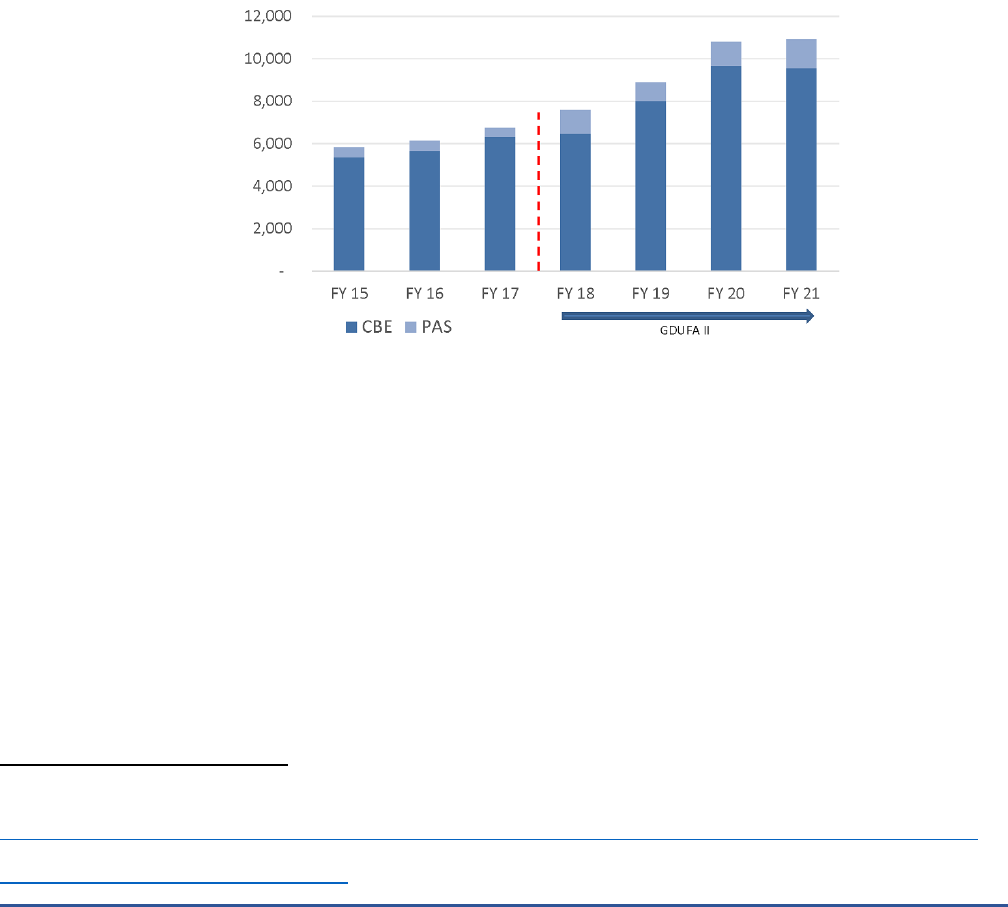
Once a generic drug application has been approved by FDA, any changes must be reported to FDA. One
of the goals of GDUFA was to reduce the review time of these change requests. Prior-approval supplements
(PAS) are required for any major change that has a substantial potential to have an adverse effect on the
identity, strength, quality, purity, or potency of a drug product as these factors may relate to the safety or
effectiveness of the drug product; a moderate change requires a “changes being effected” (CBE) notification.
The number of PAS and CBE submissions has almost doubled since GDUFA was first passed (Figure 4).
24
Figure 4. Generic Drug Applications for Prior-Approval Supplements (PAS) and Changes Being Effected (CBE)
Supplement
24
Notes: The dashed red line represents passage of GDUFA II. “PAS” is an abbreviation for “prior-approval supplements”, which are required
by FDA for applicant holders to submit prior to making any major change to a generic drug. “CBE” is an abbreviation for “changes being
effected” supplements, which are required by FDA for applicant holders to submit when making moderate changes to a generic drug.
EVIDENCE ON USER FEES
ASPE conducted literature reviews, including ASPE-generated research and academic research, to
identify empirical studies on several topics: the role of user fees on drug prices, the share of the costs for
medical product development spent on user fees, and the impact of user fees on review timelines.
Relationship between User Fees and Drug Pricing
In 2021, gross prescription drug spending in the United States was $603 billion,
28
while FDA collected
$1.153 billion in prescription drug user fees.
29
This means that, in aggregate, user fees made up 0.20 percent of
realized revenue for prescription drugs in 2021. Similarly, for medical devices, the most recent spending
28
Parasrampuria, S. & Murphy, S. Trends in Prescription Drug Spending, 2016-2021. Washington, DC: Office of the Assistant
Secretary for Planning and Evaluation, U.S. Department of Health and Human Services. September 2022.
https://aspe.hhs.gov/sites/default/files/documents/88c547c976e915fc31fe2c6903ac0bc9/sdp-trends-prescription-drug-spending.pdf
29
U.S. Food and Drug Administration. FY 2021 Financial Report to Congress for the Prescription Drug User Fee Act of 1992.
https://www.fda.gov/media/156199/download

March 2023 Issue Brief 8
estimates are from 2019, when spending on medical devices was $199.1 billion.
30
In 2019, FDA’s collection of
medical device user fees equaled $208 million, or 0.1 percent of the revenue for medical devices. While these
numbers are aggregates, it suggests that the market for medical products remains large and that user fees make
up a small portion of the expected revenues (well under 1 percent in both drug and medical device markets).
This, in turn, likely means that user fees are not commonly a driving factor when making decisions about
bringing products to market or the pricing of products.
We did not find any papers linking user fees to high prices of brand drugs. We identified two studies
related to generic drug user fees that could indirectly shed light on this issue via changes in market
concentration or market exit, but neither study provides direct quantitative evidence to support the relationship
between user fees and drug prices generally or for brand name or generic drugs.
31,32
Berndt et al. (2018)
assessed the structure of the GDUFA I and concluded that it created barriers to entry for new generic drug
manufacturers. Dong et al. (2017) found that GDUFA I disproportionately burdened small and new firms and
that it favored large firms. This study also suggested that high market concentration and dependence on foreign-
supply sources can increase the vulnerability of the supply chain and potentially lead to product shortages and
price changes.
33
Neither of these studies directly examined drug prices, and they examined a user fee structure
that was replaced by GDUFA II to ameliorate some of the concerns raised by these studies.
User Fees as a Share of the Total Cost of Development
ASPE has completed several research projects that examined the cost of development for different
types of medical products. The research examined the cost of development for new drugs, generic drugs,
complex medical devices, and vaccines. One of the primary goals in each of these analyses was to examine
drivers and barriers to medical product development, including the contribution of FDA user fees to the cost of
development.
30
Donahue, G.F. (2021) Estimates of Medical Device Spending in the United States. Report for Advanced Medical Technology Association.
https://www.advamed.org/wp-content/uploads/2021/12/Estimates-Medical-Device-Spending-United-States-Report-2021.pdf
31
Berndt, E. R., Conti, R. M., & Murphy, S. J. (2018). The Generic Drug User Fee Amendments: An Economic Perspective. Journal of Law
and the Biosciences, 5(1), 103-141.
32
Dong, K., Boehm, G., & Zheng, Q. (2017). Economic Impacts of the Generic Drug User Fee Act Fee Structure. Value in Health, 20(6), 792-
798.
33
Lund, S., Manyika, J., Woetzel, J., Barriball, E., Krishnan, M., et al. Risk, Resilience, and Rebalancing in Global Value Chains. McKinsey
Global Institute, August 2020.

March 2023 Issue Brief 9
ASPE developed an analytical model of medical product development using data from public and
proprietary sources with coverage from 2000 to 2018. The model estimated the cost, duration, and phase
transition success probability associated with each stage of development, including FDA review and approval.
The results of the analytical model provided
an estimate of the cost of medical product
development at each stage of the process
from the nonclinical stage to post-marketing.
In this model, the cost of the FDA review
stage was estimated using user fees paid by
industry and the average time it takes for FDA to review a marketing application. The duration of product
development varied across medical products, with some drugs taking up to 10 years to develop, and other drugs
not reaching the marketing authorization stage. These estimates considered the time it takes to develop a
medical product (i.e., “development costs”) and the likelihood that each product was successful at each stage of
the development process. That is, these costs included the cost of investment and the cost of failures also
known as “capitalized costs.” Our results show that FDA user fees made up approximately 1.0 percent of the
total capitalized cost of development for drugs,
34
2.0 percent of the total capitalized cost of development for
preventive vaccines, and 0.5 percent of the total capitalized cost of development for complex medical
devices.
35,36
In a related report from 2022,
37
ASPE developed an analytical framework that examined the expected
profit for a generic drug developer in different size drug markets. The framework included characteristics of the
type of drugs, the opportunity cost of capital, the fifteen stages of generic drug development, and future
revenue expectations. This framework formed the basis for an operational model that can simulate expected
changes in the cost of developing a generic drug based on different policy changes, such as a change in user
fees.
38
This analysis found that the average cost to develop a generic drug was $2.4 million ($3.2 million in 2022
dollars) and that it required just under 5 years to bring a generic drug to market. The average capitalized cost
was $5.3 million ($7.06 in 2022 dollars). FDA user fees to submit an ANDA constituted 1.7 percent (range: 0.2 to
7.0 percent) of expected capitalized costs.
The analysis also assessed the impact of a hypothetical 50 percent decrease in ANDA submission fees. A
50 percent decrease in ANDA submission fees would result in a 1.2 percent (range: 0.1 to 4.8 percent) decrease
in expected capitalized costs.
39
In contrast, other factors would have a much larger impact on reducing the cost
34
In our model, drugs include biologics except preventive vaccines which are modeled separately.
35
Sertkaya, A., DeVries, R., Jessup, A., Beleche, T. (2022) Estimated Cost of Developing a Therapeutic Complex Medical Device in the US.
JAMA Network Open, 5(9): e2231609. Doi: 10.1001/jamanetworkopen.2022.31609.
36
Therapeutic complex medical devices are defined as a Class III device that usually sustains or supports life, is implanted, or presents
potential unreasonable risk of illness or injury and requires a PMA application to obtain marketing approval in the U.S. These types of
applications make up less than 1 percent of all medical device submission and, as such, the results cannot be generalized to other medical
devices Further, there is variability in terms of complexity of medical products, which can result in a large degree of variability around the
average development cost.
37
Sertkaya, A., Lord, A., & Berger, C. (2022). Cost of Generic Drug Development and Approval. Report for the Office of the Assistant
Secretary for Planning and Evaluation, U.S., Department of Health and Human Services. https://aspe.hhs.gov/reports/cost-generic-drugs
38
Broadly speaking, these stages captured the initial R&D phases, in vivo testing, bridging studies, patent challenge and litigation, and
FDA review. See Sertkaya et al (2022) for additional details.
39
The hypothetical model assumes that the fee decreases would not affect FDA’s capacity to review applications in a timely manner.
Sertkaya et al (2022) note that “a fee decrease of that magnitude could severely hamper FDA’s ability to meet its congressionally
mandated review timelines and would likely increase the average FDA ANDA review time estimated in the model. This could potentially
countervail the cost-saving effect of FDA ANDA review fee reductions to the generic drug applicant and can even result in an increase in
the overall expected capitalized development cost.”
User fees made up less than 2.0 percent of the
total capitalized cost of development for drugs,
complex medical devices and preventive vaccines.

March 2023 Issue Brief 10
of generic drug development, such as increasing the rate of FDA first-cycle approvals, expanding the use of
biowaivers in lieu of bioequivalence studies where possible, removing the incentive for reference product
sponsors to “product hop,”
40
and reforming payment incentives, such as ensuring that generic drugs are placed
on preferred formulary tiers.
Taken together, ASPE research suggests that user fees are a small contributor towards the capitalized
cost of medical product development, on the order of 0.5 to 2 percent.
Impact of User Fees on FDA Review of Applications
Several studies have assessed the impact of funding on review times. Carpenter et al. (2003) examined
whether the source of funding influenced review times with a goal of addressing concerns about pharmaceutical
industry influence in the review process.
41
The authors found that user fees shortened New Drug Application
(NDA) review times by 3.3 months for every 100 additional staff and concluded that the amount of funding for
FDA staff was more influential in shortening NDA review time than the source of funding.
41
Kaitin (1997) also
found that NDA review times were shortened after PDUFA, but that user fees did not affect the overall timeline
to conduct clinical studies and hence also did not largely affect the overall timeline to bring a medical product to
market.
42
Darrow et al. (2020) found that PDUFA fees increased from $66 million ($115 million in 2022 dollars)
over the period of 1993-1997 to $820 million ($935 million in 2022 dollars) in 2013-2017 and estimated that in
2018 user fees accounted for approximately 80 percent of the salaries of drug application reviewers.
43
This same
study estimated that FDA review times declined from more than 3 years in 1983 to less than 1 year in 2017, and
that FDA accepted more surrogate endpoints and less data in their applications.
43
As a result, the authors
estimated that legislation from 1983 to 2018, including user fees, increased the number of new drug approvals.
The mean annual number of new drug approvals, including biologics, increased from 34 in 1990-1999 to 41 in
2010-2018.
43
The median annual number of generic drugs approved increased from 284 prior to GDUFA to 588
between 2013 and 2018.
43
More recent data show that in fiscal year 2021, FDA approved 579 ANDAs and
tentatively approved 157 ANDAs.
44
An older study conducted by Berndt et al. (2004) estimated that
approximately two-thirds of the decline in approval times can be attributed to PDUFA.
45
A study by Mitchell et
al. (2022) concluded that the PDUFA model supported the implementation of a range of changes to FDA policy
and statutory changes that included promoting the expedited review programs (e.g. priority review and the
breakthrough and fast track designation); increased opportunities for industry meetings with FDA about clinical
trial design; guidance on rules regarding industry dissemination of peer reviewed scientific literature and other
health care economic information to providers; and incentives to encourage market entry of new generic drugs.
The authors suggest that FDA’s budgetary dependence on user fees and industry’s required participation in
PDUFA negotiations may advantage the industry.
46
40
A “product hop” refers to a practice where a brand company attempts to preserve its market dominance by creating a new version of a
product with incremental innovation that is protected by a longer-term patent.
41
Carpenter, D., Chernew, M., Smith, D. G., & Fendrick, A. M. (2003). Approval Times for New Drugs: Does The Source Of Funding For FDA
Staff Matter? The Amount of Resources Devoted to FDA Review, Not the Source of Funding, Was Likely the Principal Driver Behind
Shrinking Approval Times Since 1980. Health Affairs, 22(Suppl1), W3-618.
42
Kaitin, K. I. (1997). The Prescription Drug User Fee Act of 1992 and the New Drug Development Process. American Journal of
Therapeutics, 4(5-6), 167-172.
43
Darrow, J. J., Avorn, J., & Kesselheim, A. S. (2020). FDA Approval and Regulation of Pharmaceuticals, 1983-2018. JAMA, 323(2), 164-176.
44
U.S. Food and Drug Administration. FY 2021 Performance Report to Congress for the Generic Drug User Fee Amendments.
https://www.fda.gov/media/155760/download.
45
Berndt, E. R., Gottschalk, A. H., Philipson, T., & Strobeck, M. W. (2005). Assessing the Impacts of the Prescription Drug User Fee Acts
(PDUFA) on the FDA Approval Process. In Forum for Health Economics & Policy (Vol. 8, No. 1). De Gruyter.
46
Mitchell, A. P., Trivedi, N. U., & Bach, P. B. (2022). The Prescription Drug User Fee Act: Much More Than User Fees. Medical Care, 60(4),
287-293.

March 2023 Issue Brief 11
Economic Benefits of User Fees
User fees help support allowable activities that have implications for patient safety and access to new
medical products. For example, user fees can support increased staffing and training to speed up review of NDA
and Biologics License Applications (BLA) manufacturing supplements. NDA or BLA manufacturing supplements
are submitted to FDA when a manufacturer intends to make post-approval changes to manufacturing sites,
manufacturing processes, or other changes that are considered a major change.
47
A major change is one that
has a substantial potential to have an adverse effect on the safety and efficacy of the approved product and
must be approved by FDA before it is made. Further, additional staffing can help bring medical products to
market more expeditiously and thereby increase medical care options for patients.
Putting all of these various effects together, a few studies have examined the impact of medical product
user fees, including the economic benefits of user fees. One of the few studies that provides quantitative
estimates of the economic benefits of PDUFA on society is a study by Philipson et al. (2008), which found that
PDUFA resulted in net benefits for consumers and manufacturers. Specifically, the authors showed that PDUFA
raised manufacturers’ returns on investment by $7 to $11 billion ($9.3 to $14.7 billion in 2022 dollars) or about
$25 to $39 million per drug launched ($33.3 to $52.0 million in 2022 dollars).
48
These benefits were mainly
driven by reduced review times, which accelerated the launch of new drugs to market and hence to patients.
These estimates suggest that for $1 paid in user fees, the return for manufacturers is between $8.60 and $13
($11.5 and $17.3 million in 2022 dollars). Philipson et al. (2008) also showed that consumer welfare as measured
by the health benefits from access to more rapid approval of drugs under PDUFA increased between $7 billion
and $20 billion ($9.3 and $26.6 billion in 2022 dollars).
48
These estimated benefits are substantial in comparison
to user fees (about $3 million per NDA submission or $1.8 billion in total user fees for devices, human drugs and
biologics authorized in FY2022). Further, these estimates can also be translated into 140,000 to 310,000 life
years saved from patients who have more rapid access of drugs.
48
The authors concluded that the estimated net
benefits far outweighed the estimated 56,000 life years lost due to adverse events related to the use of
approved drugs that were subject to PDUFA fees and later withdrawn for safety reasons.
48
It is important to
note that this paper was published 15 years ago and there have been significant changes in the prescription drug
marketplace and landscape in the intervening period, so further examination of whether these estimates still
hold would be beneficial.
Discussion and Conclusion
The existing body of work points to benefits to consumers and industry from user fees in the form of
increased access to new medical products, faster review timelines, and improvements in the transparency of the
regulatory review process. The evidence also suggests that these benefits outweigh the costs, as user fees
represent a small percent of development costs and serve as a crucial source of funding for regulatory activities
and approval of medical products. User fees account for 66 percent of the human drugs budget, 43 percent of
the biologics budget, and 35 percent of the medical devices budget.
Most of the existing research on user fees has focused on the largest user fee programs for prescription
drugs and generics; however, examining the impact of user fees for biosimilars and medical devices may provide
new insights. This is particularly important in light of the COVID-19 pandemic during which there has been an
47
FDA’s regulations also require submission of a supplement for certain other than major changes. For example, a supplement
submission is required at least 30 days prior to distribution of the product made using the change.
48
Philipson, T., Berndt, E. R., Gottschalk, A. H., & Sun, E. (2008). Cost-benefit Analysis of the FDA: The Case of the Prescription Drug User
Fee Acts. Journal of Public Economics, 92(5-6), 1306-1325

March 2023 Issue Brief 12
unprecedented increase in the number of medical device submissions to FDA. As the pace of technological
development continues to increase, new medical products will continue evolving and become more complex.
This in turn requires having adequate funding and resources to conduct regulatory activities and reviews
necessary to keep pace. However, given that some of the key studies are 20 years old, there could be potential
value in updating the research on user fees in recognition of the changing marketplace and landscape.

March 2023 Issue Brief 13
U.S. DEPARTMENT OF HEALTH AND HUMAN SERVICES
Office of the Assistant Secretary for Planning and Evaluation
200 Independence Avenue SW, Mailstop 434E
Washington, D.C. 20201
For more ASPE briefs and other publications, visit: aspe.hhs.gov/reports
ABOUT THE AUTHORS
Sonal Parasrampuria is a Social Science Analyst and FDA portfolio lead in the Office of Science and Data Policy in ASPE.
Trini Beleche is a Senior Economist in the Office of Science and Data Policy in ASPE.
SUGGESTED CITATION
Parasrampuria, S., and Beleche, T.
FDA User Fees: Examining Changes in Medical Product Development and Economic
Benefits. Washington, DC: Office of the Assistant Secretary for Planning and Evaluation, U.S. Department of Health and
Human Services. March 2023.
COPYRIGHT INFORMATION
All material appearing in this report is in the public domain and may be reproduced or copied without permission; citation
as to source, however, is appreciated.
___________________________________
For general questions or general information about ASPE: aspe.hhs.gov/about
